

El genotipo F del ecovirus 25 con patrón de recombinación múltiple ha estado circulando de forma continua y amplia en China continental.
Se reclasificaron nueve genotipos en función de las secuencias de VP1.
Según las pautas de clasificación de genotipos, las diferencias de nucleótidos oscilan entre el 15 y el 25% entre genotipos y menos del 15% dentro de los genotipos.dieciséis, 113 secuencias de VP1 de longitud completa de E25 se pueden clasificar en nueve genotipos (A – I) (Figura 1). Las diferencias de nucleótidos entre los nueve genotipos oscilaron entre 15,7 y 24,7%, y en la Tabla 1 se dan detalles sobre las diferencias de nucleótidos y aminoácidos entre los diferentes genotipos.
La máxima probabilidad del árbol filogenético depende del conjunto. Vicepresidente1 Genoma de 113 secuencias del genoma E25. círculo lleno: ⚫Cepas E25 en este estudio. Rombo relleno: ◆Cepa modelo E25.
La cepa prototipo (JV-4) se designó como genotipo A y los ocho genotipos restantes se nombraron cronológicamente. Los genotipos B, C e I, que constan de una sola secuencia, se aislaron en Australia en 1995, China en 1998 y Estados Unidos en 2016, respectivamente. El genotipo D incluye 22 secuencias, la mayoría de las cuales (16/22) fueron aisladas en la India entre 2010 y 2012. Asimismo, el genotipo H estuvo compuesto principalmente por cepas indias (6/8) y dos chinas. El genotipo E 6 incluye secuencias de Australia, Alemania, Taiwán y China. Las cinco secuencias del genotipo G procedían de Australia, Francia y Estados Unidos. El genotipo F incluía 68 secuencias que abarcaban cuatro países, a saber, China, EE. UU., Australia y Francia, de las cuales 55 secuencias eran de China, y este genotipo era también el principal genotipo dominante en China continental.
Análisis dinámico de E25.
Utilizando el software BEAST, se analizaron 110 VP1 E25 de longitud completa para determinar su origen evolutivo. Los resultados mostraron que la tasa de evolución promedio para E25 fue 6.08 × 10-3 Alternativas/Ubicación/Año (95% HPD: 5.10 x 10-3 a 7,16 x 10-3); La época de origen fue 1923 (95% HPD: 1901 a 1944) (Tabla 1, Figura 2A). Por el contrario, el genotipo F surgió alrededor de 1993 con una tasa evolutiva de 6,44 × 10-3 Alternativas/Ubicación/Año (95% HPD:5.53 x 10-3 a 7,34 x 10-3) (Tabla 1). El gráfico del horizonte bayesiano muestra que el tamaño de la población E25 fue estable y constante hasta 2004, con pequeñas fluctuaciones crecientes y decrecientes de 2004 a 2008; Luego, hubo un ligero aumento después de que comenzó a disminuir de 2012 a 2016; y un estado estable después de 2016 (Figura 2B). El genotipo F mostró poca fluctuación entre 2008 y 2014 (Figura 2C). Sin embargo, las secuencias de otros genotipos no son adecuadas para el análisis con BEAST solo debido a los grandes errores causados por el pequeño número de secuencias.
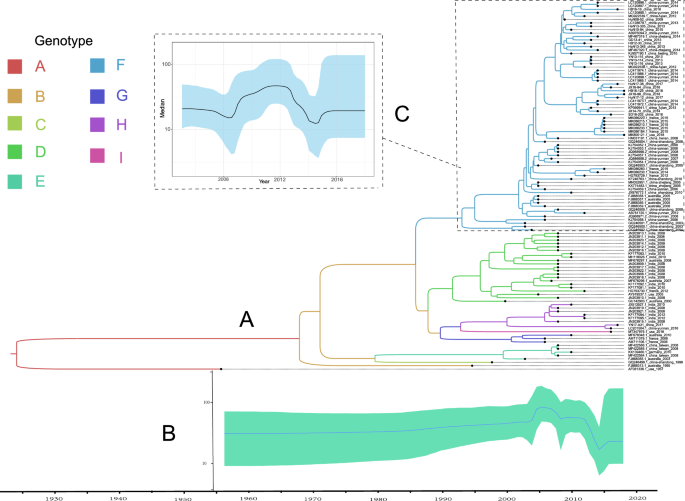
(a) El árbol filogenético de máxima credibilidad (MCC) se construyó utilizando el método Monte Carlo de la cadena de Markov (MCMC) basado en 110 secuencias completas de VP1 de E25. El color de las ramas representa aislados de diferentes genotipos. (B) Gráfico del horizonte bayesiano de la secuencia de toda la región VP1 de E25, que refleja la diversidad genética relativa de E25 de 1957 a 2018. El eje X es la escala de tiempo (año) y el eje Y es el tamaño efectivo de la población; La línea continua es la media estimada y el tono azul es la intensidad de fondo más alta del 95 %. (C) Gráfico del horizonte bayesiano de la secuencia de la región VP1 del genotipo E25 F.
Seis importantes vías de transporte geográfico global para el E25
A partir de las 110 secuencias VP1 completas de E25 mencionadas anteriormente, se construyó una base de datos de secuencias que incluye seis países (EE. UU., China, Australia, Francia, Alemania e India) para analizar la dinámica espaciotemporal global de E25. Con base en un factor de Bayes (BF) ≥ 10 y una probabilidad posterior (PP) ≥ 0,5, se identificaron seis vías de transmisión significativas: de China a Alemania (BF = 23,88, PP = 0,85); China a Francia (BF = 37,23, PP = 0,90); China a Australia (BF = 21,56, PP = 0,83); India a EE.UU. (BF = 12,24, PP = 0,74); India a Australia (BF = 400,25, PP = 0,99); y Francia a EE. UU. (BF = 12,17, PP = 0,74) (Figura 3A, Tabla complementaria S3). Las trayectorias anteriores muestran que el E25 se propaga principalmente desde Asia al resto del mundo. Además, los resultados de la recompensa de Markov mostraron que China domina la producción de E25 en todo el mundo con un valor de recompensa de Markov de 11,32, que es mucho más alto que el de otros países (Figura 3b, Tabla complementaria S4). Sin embargo, los resultados están algo sesgados debido al número limitado de series globales disponibles para cada país.

(a) Ruta de transporte espacial global para E25. (B) El gráfico del número promedio de transiciones de países se basa en la ubicación geográfica de seis países.
Análisis de patrones de recombinación de E25.
Para comprender mejor el tipo recombinante de E25, se utilizaron un total de 37 secuencias que contienen 7 genotipos (A, D – I) para construir árboles filogenéticos basados en las regiones P1, P2 y P3 con prototipos para otros serotipos en el EVB. grupo, respectivamente. Entre ellas, se obtuvieron 18 secuencias de este estudio y se descargaron 19 secuencias de GenBank (Figura 4A-C). Los resultados mostraron que las 37 cepas E25 se agruparon en la región P1 y se ramificaron en las regiones P2 y P3. De acuerdo con la posición de secuencias con diferentes genotipos en el árbol filogenético en las regiones P2 y P3, determinamos que si las diferencias de nucleótidos entre diferentes cepas son superiores al 15%, se pueden reconocer como linajes de recombinación válidos. Identificamos 18 cepas (incluida una cepa modelo) para facilitar el análisis de sus patrones de recombinación (Figura 4A-C, Tabla complementaria S5), y el genotipo F podría dividirse en nueve linajes (linaje F1-F9), mientras que el genotipo E linaje único ( linaje e). El análisis utilizando el software Simplot reveló diferencias significativas en la similitud de nucleótidos entre el genotipo E y el genotipo F, y las nueve cepas del genotipo F mostraron diferencias significativas en las regiones P2 y P3 (Figura 4D). Esto también es consistente con los resultados de los árboles filogenéticos generados por P2 y P3 y también muestra que el patrón de recombinación varía entre diferentes linajes. Según las diferencias entre estos linajes se han identificado 17 patrones de recombinación, de los cuales el genotipo F se puede dividir en 9 patrones de recombinación. Para una mayor validación, seleccionamos aleatoriamente una cepa de cada uno de estos 17 linajes como secuencia de referencia y utilizamos el software RDP4 para el análisis de recombinación, que mostró que la información de la posición del punto de interrupción para cada una de las 17 secuencias de referencia era diferente, así como los serotipos. . Las cepas dominantes combinadas con las 17 cepas de referencia también difirieron significativamente (Figura 4E, Tabla complementaria S6). Esto también confirma que los patrones de reorganización de los 17 linajes clasificados son efectivamente diferentes.
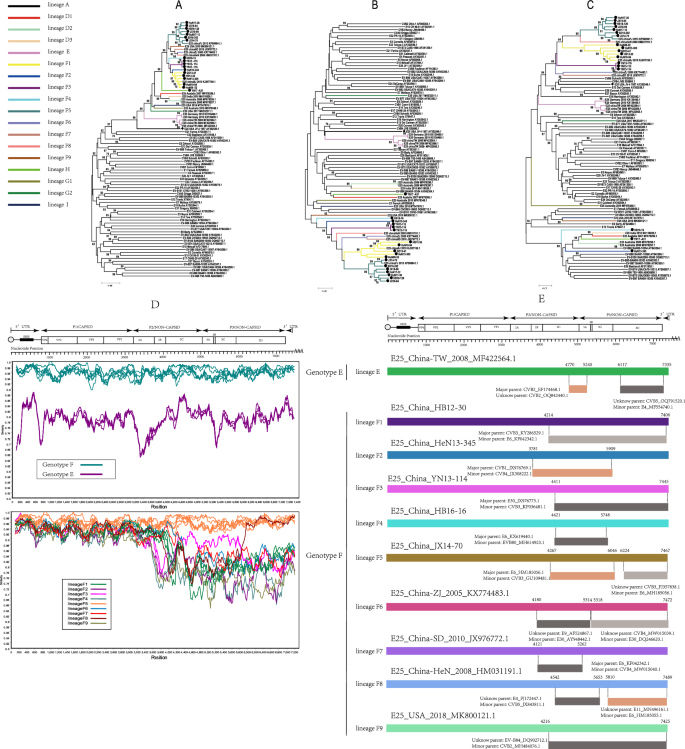
Los árboles NJ se basan en las regiones P1, P2 y P3 de las secuencias prototipo de todos los enterovirus B en la base de datos GenBank con 37 cepas E25. Diamante relleno: indica la cepa prototipo E25 (JV-4), círculo relleno: cepas E25 en este estudio. Los números en los íconos indican el soporte de arranque del nodo (frecuencia de arranque 1000). (a) secuencias codificantes de P1; (B) secuencias codificantes de P2; (C) pág.3 Secuencias de codificación. (Dr) Puntos de interrupción de recombinación basados en genomas completos de diferentes linajes detectados dentro de los genotipos. (h) Mapeo del genoma de eventos de recombinación de linaje predichos representativos utilizando el programa RDP4 para E25.
Se detectó un sitio de selección positiva en el genotipo F.
Debido a que las secuencias de la región P1 de otros genotipos eran raras o estaban ausentes (Tabla complementaria S6), seleccionamos el genotipo F, que tiene una cantidad relativamente grande de secuencias, para el análisis. Al analizar la presión global de selección del genotipo F a nivel mundial, encontramos que la proporción promedio de sustituciones de aminoácidos no sinónimos y sinónimos (dN/dS) en la región P1 fue de 0,207. Hubo un sitio de selección positiva en la posición del aminoácido 274 en la región VP1, que fue identificado por los modelos MEME y SCLA. Además, los aminoácidos en este sitio también diferían entre las cepas de referencia en las nueve cepas diferentes del genotipo F (Figura 5).
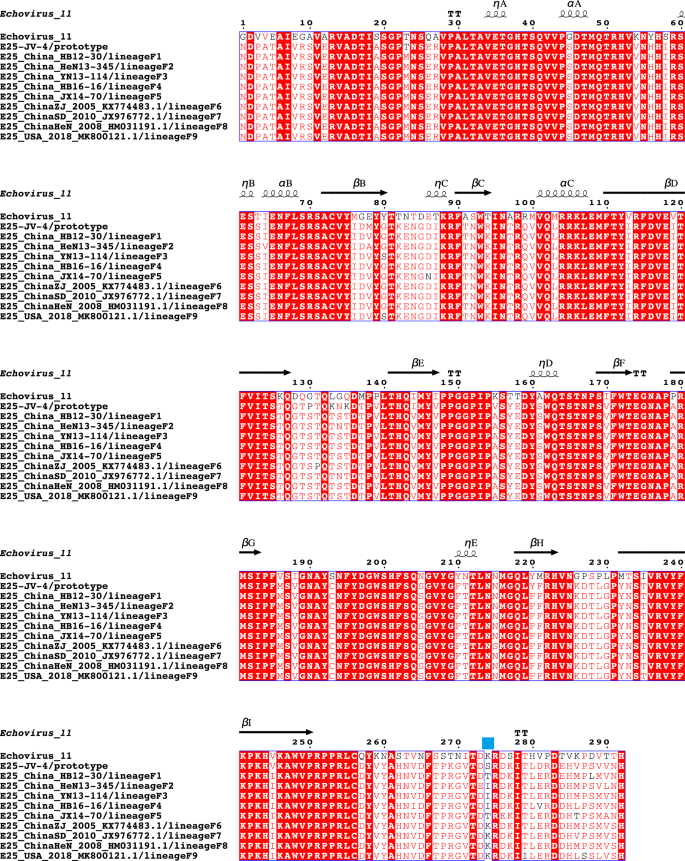
Sitios de identificación de aminoácidos positivos en la región VP1 en E25. Los sitios de selección positiva están ubicados en las marcas cuadradas azules.

“Defensor de la Web. Geek de la comida galardonado. Incapaz de escribir con guantes de boxeo puestos. Apasionado jugador”.