

Mayor tasa de detección de amplicón fuera del objetivo en MiSeq v3 en comparación con los juegos de reactivos v2 en el contexto de la secuenciación de 16S-rRNA
Primero, nuestro objetivo fue determinar el alcance de la amplificación fuera del objetivo en muestras nasofaríngeas. Uso del método de purificación AMPure XP en combinación con el kit de reactivos Miseq v310identificamos un total de 8978 ASV, de los cuales 577 (6,4 %) estaban fuera del objetivo (es decir, longitud de lectura <250 أو> 256 nt). Al integrar el número/abundancia de lecturas de ASV, estos ASV fuera del objetivo corresponden al 13,9 % de todas las lecturas (1,7 M/12,1 M de lecturas). Validamos nuestra definición basada en la longitud de las lecturas fuera del objetivo mostrando que el 99,5 % (norte= 1,661,316) están alineados con el genoma humano, lo que indica un origen humano (como mitocondrial) en lugar de bacteriano. Entre estas lecturas, el 99,8% (norte= 1 658 036 lecturas) tenían una longitud de 200 nucleótidos, que es mucho más corta en comparación con las lecturas objetivo (250–256 nm). Además, encontramos una pequeña fracción de lecturas (0,5 % de todas las lecturas fuera del objetivo; norte= 8428 lecturas) no se puede mapear en el genoma humano. Aunque también fuera del objetivo (es decir, no dentro del rango de longitud de lectura de 250 a 256), estas lecturas representan un conjunto muy dispar de lecturas, con el 99,0 % de estas lecturas de más de 200 nucleótidos (rango de 203 a 330 nm). Por lo tanto, asumimos que estas lecturas de baja abundancia representan binarios de cebadores, errores de secuenciación y fusiones fallidas de lecturas directas e inversas.
El trabajo anterior de Locker et al. ya demostró que la amplificación fuera del objetivo es un problema al secuenciar 16S-rRNA usando los cebadores V3-V4 para determinar la composición microbiana de muestras de biopsias humanas invasivas, ya que el ADN humano puede ser >97%7. En nuestro estudio, encontramos que la amplificación fuera del objetivo también ocurre con los cebadores V4 cuando se secuencian muestras de mucosas respiratorias no invasivas (AMPure XP/v3; media de 13,3 % de lecturas fuera del objetivo por muestra, rango 0,03-72,2 %). Además de la literatura anterior, pudimos demostrar una relación inversa logarítmica entre la densidad bacteriana y la proporción de lecturas fuera del objetivo (Pearson). s– 0.671, s– Valor 7,79 x 10– 30; Figura 2), lo que indica que el grado de amplificación fuera del objetivo está relacionado con la densidad bacteriana. Además, encontramos un fuerte aumento en la detección de lecturas fuera del objetivo en muestras con una densidad bacteriana inferior a 0,3 pg/μL, lo que indica que la amplificación de las lecturas fuera del objetivo es particularmente problemática en muestras con una densidad bacteriana por debajo de este umbral. Se encontró una relación similar para la mezcla basada en gel/v3 (Pearson s– 0.665, s– Valor 3,67 x 10– 29, Figura complementaria S1). Especulamos que la baja biomasa bacteriana a su vez puede estar asociada con una alta proporción de ADN de huésped a bacteriano, lo que contribuye aún más a la generación de amplicones fuera del objetivo.7. Esto se verificó al mostrar que la amplificación de lectura fuera del objetivo es rara en los controles negativos (extracción y PCR), lo que indica que el ADN mitocondrial/humano debe estar presente para que se produzca la amplificación de lectura fuera del objetivo (Figura complementaria S2). Además, establecimos una correlación negativa entre la puntuación de calidad media de las lecturas inversas (media de todos los nucleótidos) y la puntuación de detección fuera del objetivo (Pearson). s– 0.392, s– Valor 1,94 x 10– 9 Figura complementaria S3), probablemente como resultado de la asociación conocida entre la densidad bacteriana y la calidad de lectura.
Relación lineal inversa entre logaritmos y registros entre la densidad bacteriana y la tasa de detección de amplicón fuera del objetivo. El área sombreada alrededor de la línea negra representa el intervalo de confianza del 95 %. Los datos se generaron utilizando la purificación de la biblioteca AMPure XP/el conjunto de reactivos MiSeq v3 (norte= 214 muestras faríngeas). La línea horizontal roja indica el umbral en el que la detección fuera del objetivo aumenta significativamente.
A continuación, exploramos el efecto de la matriz de detectores MiSeq en la detección de amplicones fuera del objetivo. Este efecto fue sorprendentemente fuerte, mostrando una media del 0,1 % (rango 0,0-1,2 %) en comparación con el 13,3 % de lecturas fuera del objetivo por muestra para v2 y v3, respectivamente (resultados basados en AMPure XP; modelo lineal de efectos mixtos; s– Valor 1,1 x 10– 50; higos. 3). La mayor tasa de detección de amplicones fuera del objetivo se mitigó parcialmente con el kit de reactivos MiSeq v3 que utiliza un método de purificación de biblioteca basado en gel en lugar de solo AMPure XP, lo que resultó en un promedio de 4,7 % de lecturas fuera del objetivo detectadas en todas las muestras (rango) 0,0 – 39,4% ; s– Valor 2,8 x 10– 31; higos. 3). Esto probablemente se deba a una extracción y purificación más precisas de fragmentos de ADN de tamaño objetivo que otros fragmentos (de un solo tamaño) mediante la purificación basada en gel en comparación con AMPure XP.19. Sin embargo, descubrimos que el porcentaje de lecturas fuera del objetivo detectadas fue significativamente mayor para el grupo v3 en comparación con v2 cuando se usaba el método de purificación basado en gel en lugar del método de purificación AMPure XP (s– Valor 4,7 x 10– 5; higos. 3). Debido a que generamos un conjunto de muestras de amplicón de PCR en la configuración de secuenciación de MiSeq v2 y v3 (Fig. 1), pudimos capturar específicamente el efecto del conjunto de secuenciación en la detección fuera del objetivo en lugar del procedimiento de amplificación. Aunque no pudimos determinar el mecanismo exacto detrás de esta observación, especulamos que la diferencia en la detección fuera del objetivo refleja diferencias en los productos químicos o la receta de secuenciación. Hasta donde sabemos, este estudio es el primero en informar diferencias en la tasa de detección de amplicón fuera del objetivo entre el grupo v2 y v3.

Proporción de lectura fuera del objetivo en kits de reactivos MiSeq y métodos de purificación de bibliotecas. La significación se evaluó utilizando modelos lineales mixtos y Eminencia-Paquete18 para comparaciones por pares. Los diagramas de cajas representan los percentiles 25 y 75 (límites inferior y superior de las cajas, respectivamente), la mediana (línea horizontal central) y las medidas que se encuentran dentro de 1,5 veces el rango intercuartil (IQR; distancia entre los percentiles 25 y 75; bigotes) . Los medios aparecen en forma de diamante. norte= 214 puntos de datos para cada grupo/método de purificación probado.
Según Illumina, el kit detector MiSeq v3 garantiza el doble de salida de activación única (3,3-15 Gb) en comparación con el kit v2 (0,54-8,5 Gb)20. Validamos esto configurando el número total de lecturas de destino (activadas) para la matriz v3 y v2 utilizando el método de purificación AMPure XP. A pesar de la mayor detección de lecturas fuera del objetivo cuando se usa la matriz de detectores MiSeq v3 en comparación con la v2, descubrimos que después de eliminar las lecturas fuera del objetivo, la cantidad de lecturas para la matriz v3 seguía siendo aproximadamente 2 veces mayor que la v2 (promedio de veces diferencia 1,95×, rango 1,74–2,22× para muestras emparejadas, modelo lineal mixto; s– Valor 6,4 x 10– 176; Figura 4). Se encontraron resultados similares para el método de purificación basado en gel (Figura complementaria S4). Además de generar una mayor cantidad de amplicones bacterianos, la matriz de reactivos MiSeq v3 ofrece una puntuación de calidad mejorada y una mayor densidad de masa, razón por la cual la matriz v3 aún se prefiere a la v2.10.
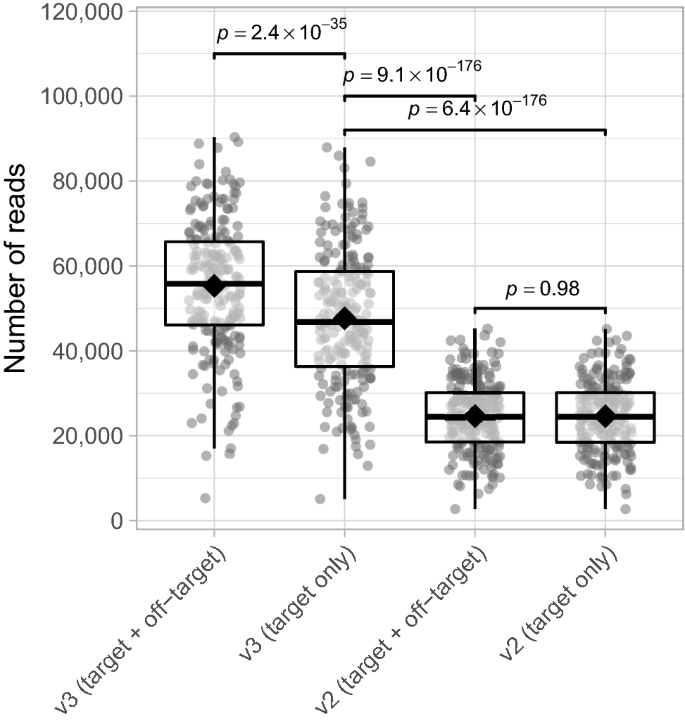
Número de lecturas (activadas)/no deseadas para los conjuntos de detectores MiSeq v2 y v3. La significación se evaluó utilizando modelos lineales mixtos y Eminencia-Paquete18 para comparaciones por pares. Ver la explicación de la Figura 3 para la definición de los elementos de un diagrama de caja. Los datos que se muestran se generaron utilizando el método de purificación AMPure XP. norte= 214 puntos de datos por grupo fotografiado.
Juntos, nuestros hallazgos demostraron que al caracterizar muestras de baja biomasa, actualmente no explotamos todo el potencial del secuenciador MiSeq cuando usamos la matriz de reactivos v3 donde también se ordena una alta proporción de lecturas fuera del objetivo. Al aplicar un método de purificación de bibliotecas basado en gel, podemos reducir la proporción de amplicones de secuencia fuera del objetivo, aunque el problema persiste hasta cierto punto. Creemos que nuestro estudio amplía los hallazgos previos de la amplificación fuera del objetivo y genera nuevos datos sobre el efecto del método de purificación de la biblioteca/matriz de reactivos en la detección de amplicones fuera del objetivo. Aunque creamos esto para el secuenciador MiSeq, no estamos seguros de hasta qué punto ocurre este problema con otros secuenciadores. Aunque nosotros y otros hemos demostrado V4-21 Y para pares de cebadores V3-V47esperamos que la amplificación de lectura fuera del objetivo pueda variar con la especificidad del cebador.
Nuestros hallazgos permiten a los investigadores anticipar la pérdida de lecturas cuando planifican estudios con muestras de biomasa baja y/o estudios que usan muestras que se prevé que tengan una alta proporción de ADN de huésped a bacteriano (por ejemplo, muestras de biopsia). Los investigadores pueden, por ejemplo, optar por agregar menos muestras a su proceso de secuenciación, incorporar una purificación de grupos de amplicones basada en gel o experimentar con pares de cebadores 16S para mantener una profundidad de secuenciación suficiente. Se debe tener especial cuidado cuando se trabaja con muestras por debajo de 0,3 pg/µL. Además, aumentamos la importancia de incorporar un paso de filtro basado en la longitud de lectura en las canalizaciones bioinformáticas utilizadas en estudios basados en 16S-rRNA para garantizar una inferencia precisa de la composición microbiana en el análisis posterior. En última instancia, nuestro estudio puede servir como punto de partida para una mayor investigación de por qué la detección de amplicón fuera del objetivo aparece más claramente en la matriz de reactivos MiSeq v3 que en v2 en muestras de baja biomasa.

“Defensor de la Web. Geek de la comida galardonado. Incapaz de escribir con guantes de boxeo puestos. Apasionado jugador”.